

Auf der sicheren Seite – Mit einem vollständigen Qualitätssicherungssystem
Unser Unternehmen verfügt über ein Medizinprodukte-Qualitätsmanagementsystem gemäss EN ISO 13485 und erfüllt ebenso die Anforderungen der Europäischen Verordnung für Medizinprodukte (kurz: MDR) und erklärt die Konformität seiner Produkte gemäss dieser Verordnung.
Unser Engagement: Für eine gesunde Gesellschaft und für Ihre rechtliche Absicherung.




Alle Produkte, die zu einem medizinischen Zweck angewendet werden, wie Diagnostik, Behandlung oder Linderung von Krankheiten. Unter anderem Ultraschallgeräte, Endoskope, Implantate und auch Trainingsgeräte in Therapie und Rehabilitation zeichnen sich durch geprüfte Genauigkeit, technischer- und biologischer Sicherheit, Wirksamkeit und intuitiver Gebrauchstauglichkeit aus. An diese Produkte werden die höchsten Anforderungen gestellt.
Die Anforderungen sind immens hoch. Im Rahmen der europäischen Medizinprodukteverordnung EU 2017/745 und eines in der Regel zertifizierten Qualitätsmanagementsystems nach EN ISO 13485 und werden alle Schritte in der Planung, Entwicklung und Produktion unter Anwendung internationaler Normen von einem Team hochqualifizierter und motivierter Experten systematisch geplant, umgesetzt, verifiziert und validiert.
Diese Anforderungen gelten auch für Hersteller mit Sitz ausserhalb der EU, wenn diese innerhalb der EU Medizinprodukte Inverkehrbringen.
In jährlichen Audits (Überwachungen) wird die Einhaltung vorgegebener Qualitätsstandards von unabhängigen Stellen (In unserem Falle der TÜV Süd) geprüft und bewertet.
Grundsätzlich ist dies auf der Konformitätserklärung für das Produkt ersichtlich. Hier gibt der Hersteller an, welche Rechtsanforderungen das Produkt erfüllt. Im Falle von Medizinprodukten mindestens die Europäische Verordnung für Medizinprodukte (2017/745 – MDR). Zudem ist der Hersteller bzw. Ihr Gerätelieferant verpflichtet, auf Anfrage zu jedem Produkt diese Konformitätserklärung bereitzustellen. Jeder Einrichtungsbetreiber sollte diese Konformitätserklärungen in seiner Einrichtung bereithalten, um im Falle von Anfragen der zuständigen Landesbehörden mögliche rechtliche Konsequenzen zu vermeiden.
An jedem Gerät muss diese Konformität mit einem CE-Kennzeichen verdeutlicht werden. In der Regel befindet sich dies auf dem Typenschild. Bei vielen Medizinprodukten (höherer Klassifizierung) muss eine vierstellige Kennnummer einer Benannten Stelle hinter dem CE-Kennzeichen angegeben sein (z. B. TÜV Süd – CE 0123).
Es gibt Medizinprodukte für die verschiedensten Anwendergruppen. Der Hersteller legt diese im Rahmen der Zweckbestimmung fest. So gibt es auch speziell für den Laienanwender bestimmte Medizinprodukte.
Für Medizinprodukte in der professionellen Anwendung, sieht der Gesetzgeber zu Recht vor, dass nur solche Therapeuten und Anwender Diagnostik und Therapie durchführen dürfen, die eine entsprechende Ausbildung absolviert haben und in die medizinischen Geräte eingewiesen wurden.
Trainingsgeräte, die als Medizinprodukte zugelassen sind, verfügen über
- eine wissenschaftlich geprüfte klinische Wirksamkeit,
- ein lückenloses Risikomanagement,
- geprüfte biologische, elektrische, mechanische Sicherheit.
Die Produkte unterliegen auch während des Einsatzes beim Anwender einer permanent technischen und klinischen Überwachung, um Risiken zu minimieren und Therapieerfolg sicher zu stellen. Ein Leben lang.
Medizinprodukte unterliegen regelmässigen, vom Hersteller definierten Sicherheits- und/oder Messtechnischen Überprüfungen bzw. Kontrollen. Die Nachweise der Überprüfungen oder Kontrollen müssen auf Anfrage den Landesämtern für soziale Dienste vorgelegt werden.
Durch den Abschluss eines Wartungsvertrages können die regelmässigen Überprüfungen automatisiert durchgeführt und lückenlos nachgewiesen werden.
Bitte beachten Sie, dass die Verbindung eines unserer Medizinprodukte mit Zubehör oder einer Trainingssteuerung, die von uns nicht autorisiert wurde, zu einem Verlust der Konformität des Produktes führen kann und das Haftungsrisiko vollständig auf Sie als Betreiber übergeht.
Änderungen an Geräten beim Kunden, zum Beispiel das Aufspielen fremder Software oder das Anbringen eines anderen Displays an einem Gerät, das bereits ausgeliefert wurde, müssen vom Hersteller vorher schriftlich autorisiert werden, ansonsten verliert dieses Gerät seine geprüfte Sicherheit! Das hätte zur Folge, dass bei eventuellen Schäden und Verletzungen der Betreiber in der vollen Haftungsverantwortung steht und es erlischt die Gewährleistung und Garantiezusage.
Unsere Kunden vertrauen weltweit auf unsere medizinischen Test-/Trainings- und Therapiesysteme und unterstützen unsere Vision einer gesunden Gesellschaft.
Mit uns sind Sie auf der sicheren Seite, denn die PHYSIOMED GROUP bietet als EN ISO 13485 zertifiziertes, führendes Medizintechnikunternehmen geprüfte und höchste Sicherheits- und Qualitätsstandards: Unsere Produkte erfüllen die Anforderungen der europäischen Medizinprodukteverordnung (MDR) und tragen das CE-Zeichen.
Als zertifiziertes Unternehmen definieren wir für unsere Produkte eine Lebensdauer, in deren Zeitspanne – nach Inverkehrbringung – kein Risiko des jeweiligen Produkts für Patienten und Anwender ausgeht. Innerhalb der Produktlebensdauer sind Hersteller z. B. auch verpflichtet, Ersatzteile für etwaige Reparaturen zur Verfügung zu stellen, denn es dürfen nur Ersatzteile verwendet werden, die der Hersteller entsprechend der Konstruktion und dem Risikomanagement bereitstellt. Wir betreiben die vorgeschriebene Marktbeobachtung, die ebenfalls innerhalb der Produktlebensdauer garantiert werden muss.
Im Rahmen des gesamten Produktlebenszyklus eines Medizinproduktes muss die Abkündigung berücksichtigt werden. Unseren Kunden möchten wir einen möglichst langen und professionellen Support für unsere Produkte liefern – mit unserem Ampelsystem, das wir neben unseren Eigenprodukten auch für unsere Handelsprodukte umsetzen.
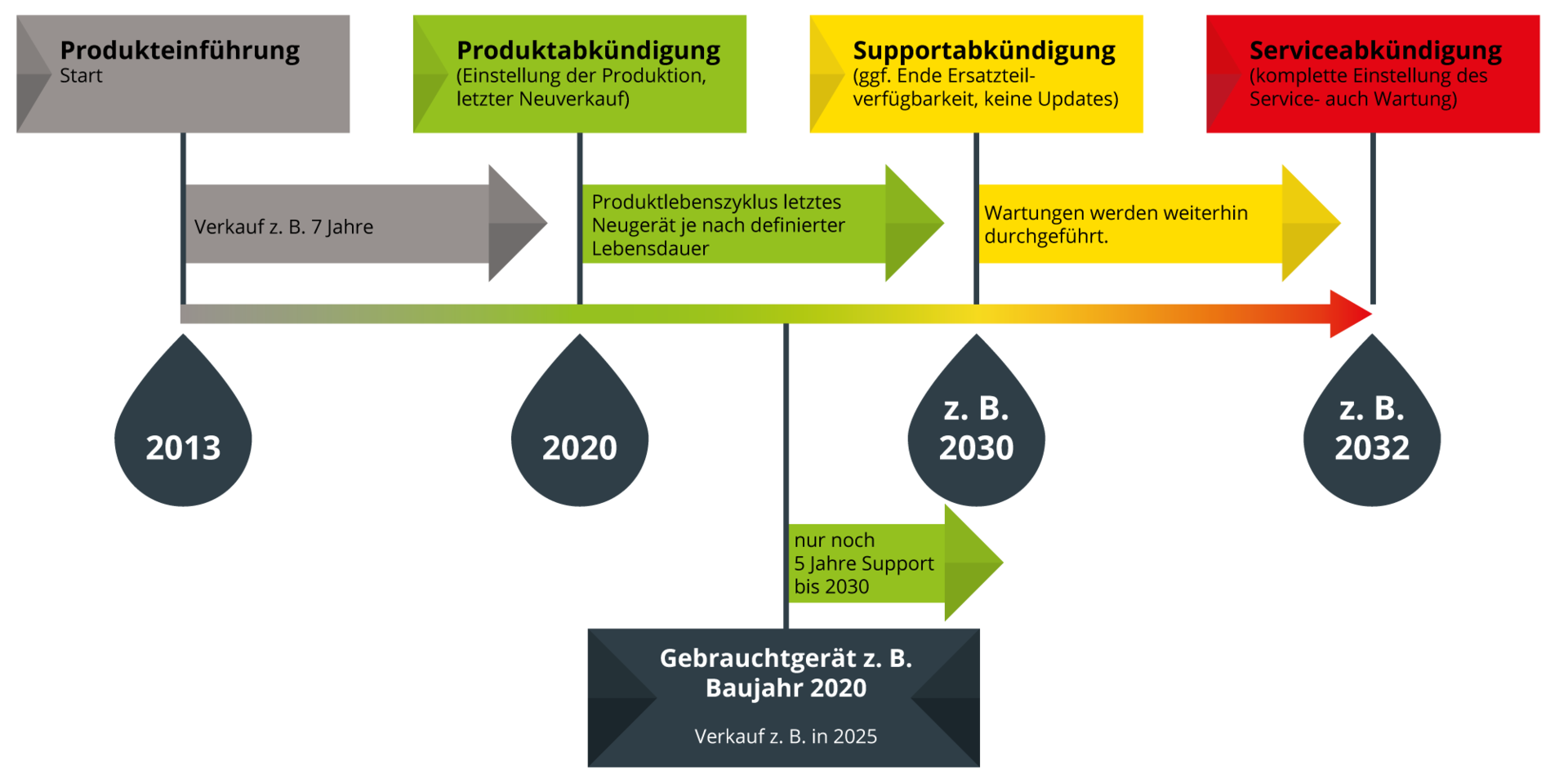
Erläuterung: Sie erwerben z. B. 2025 ein Gebrauchtgerät, Baujahr 2020. Gemäss dem Produktlebenszyklus in unserem Ampelsystem erfolgt der Support für dieses Gerät bis 2030 bis zur Supportabkündigung.
- Das Produkt wird nicht weiter produziert, es findet kein weiterer Neuverkauf statt.
- Das Produkt wird weiter supportet, Ersatzteile werden bis mindestens zum Ende der Produktlebensdauer zur Verfügung gestellt.
- Wartungen können regulär durchgeführt werden.
- Das Produkt kann weiter mit Updates/Major Updates aufgewertet werden.
- Bei Handelsprodukten: Der Hersteller kündigt das Produkt ab, was von der PHYSIOMED GROUP in dessen Namen übernommen wird, oder das Handelsprodukt wird nicht weiter über PHYSIOMED GROUP vertrieben und es wird an den Hersteller verwiesen.
- Die Produktlebensdauer des zuletzt verkauften Produktes ist abgelaufen.
- Es stehen keine oder nur noch begrenzt Ersatzteile zur Verfügung.
- Wartungen können weiterhin durchgeführt werden. Hinweis: Es wird versucht, die Funktionsfähigkeit des Systems, trotz des Alters und des möglichen Verschleisses, durch mögliche versteckte mechanische und elektrische Mängel, aufrecht zu erhalten.
- Das Produkt wird nicht weiter mit Updates/Major Updates aufgewertet (Ausnahme: Sicherheitsrelevante Updates).
- Das Produkt hat z. B. technische Mängel (z. B. Abdeckungen brechen beim Herausnehmen), oder entspricht nicht mehr dem aktuellen Stand der Entwicklung.
- Es stehen keine qualifizierten Techniker mehr zur Verfügung, die für Instandhaltungsmassnahmen nach MPG ausgebildet sind.
- Wartungen werden nicht mehr durchgeführt.
- Das Produkt wird nicht weiter mit Updates/Major Updates aufgewertet.
