

On the safe side – with a comprehensive quality assurance system
Our company operates a medical device quality management system in accordance with EN ISO 13485 and also meets the requirements of the European Medical Device Regulation (MDR), declaring the conformity of its products in line with this regulation.
Our commitment: For a healthy society – and for your legal security.




All products used for a medical purpose – such as diagnosis, treatment, or alleviation of disease. These include ultrasound devices, endoscopes, implants, and even training equipment used in therapy and rehabilitation. Such products are characterised by verified accuracy, technical and biological safety, effectiveness, and intuitive usability. They are subject to the highest standards and requirements.
The requirements are extremely high. Under the European Medical Device Regulation (EU 2017/745) and a typically certified quality management system according to EN ISO 13485, all steps in planning, development, and production are systematically planned, implemented, verified, and validated by a team of highly qualified and motivated experts using international standards.
These requirements also apply to manufacturers based outside the EU if they place medical devices on the market within the EU.
Annual audits (surveillance assessments) are conducted by independent bodies (in our case TÜV Süd) to review and verify compliance with prescribed quality standards.
This can usually be seen on the Declaration of Conformity for the product. Here, the manufacturer specifies which legal requirements the product complies with – at a minimum, the European Medical Device Regulation (2017/745 – MDR).
Manufacturers or device suppliers are also required to provide this declaration upon request. Every facility operator should keep these declarations on-site to avoid possible legal consequences in case of inspection by the relevant state authorities.
Each device must visibly display its conformity by means of the CE marking, typically found on the nameplate. For many higher-class medical devices, a four-digit identification number of a Notified Body (e.g. TÜV Süd – CE 0123) must follow the CE mark.
Medical devices are intended for a wide range of user groups, which are defined by the manufacturer within the intended purpose. Some devices are specifically designed for laypersons.
For devices intended for professional use, legislation rightly stipulates that only those therapists and practitioners who have completed appropriate training and received proper instruction in the use of the medical device are authorised to carry out diagnosis and therapy.
Training equipment certified as medical devices offers:
- Scientifically validated clinical effectiveness
- Comprehensive risk management
- Verified biological, electrical, and mechanical safety
These products are also subject to ongoing technical and clinical monitoring during their use to minimise risks and ensure successful therapy – throughout their entire lifetime.
Medical devices are subject to regular safety and/or metrological inspections, as defined by the manufacturer. Proof of these inspections or tests must be available for review by regional health authorities upon request.
By concluding a maintenance contract, these regular inspections can be automated and fully documented.
Please note: connecting one of our medical devices to accessories or training controls not authorised by us may result in a loss of conformity, and full liability will transfer to you as the operator.
Any modification to devices at the customer’s site – for example, installing third-party software or replacing the display of a delivered device – must be formally authorised in writing by the manufacturer. Otherwise, the device loses its verified safety certification!
As a result, in case of damage or injury, the operator bears full liability, and warranty and guarantee claims become void.
For more than 30 years, customers worldwide have trusted our medical testing, training, and therapy systems, supporting our vision of a healthy society.
With us, you are on the safe side: as a certified medical technology company (EN ISO 13485), PHYSIOMED guarantees the highest levels of safety and quality. Our products comply with the European Medical Device Regulation (MDR) and bear the CE mark.
As a certified company, we define a service life for our products during which, following market release, no risk to patients or users arises from the device.
Within this lifecycle, the manufacturer is obliged to make spare parts available for necessary repairs – only those parts provided in accordance with the product’s design and risk management may be used.
We also conduct mandatory market surveillance throughout the product’s lifecycle.
As part of the overall product lifecycle, the discontinuation phase must be taken into account. We aim to offer our customers the longest and most professional support possible – through our traffic light system, which we apply both to our own and to distributed products.
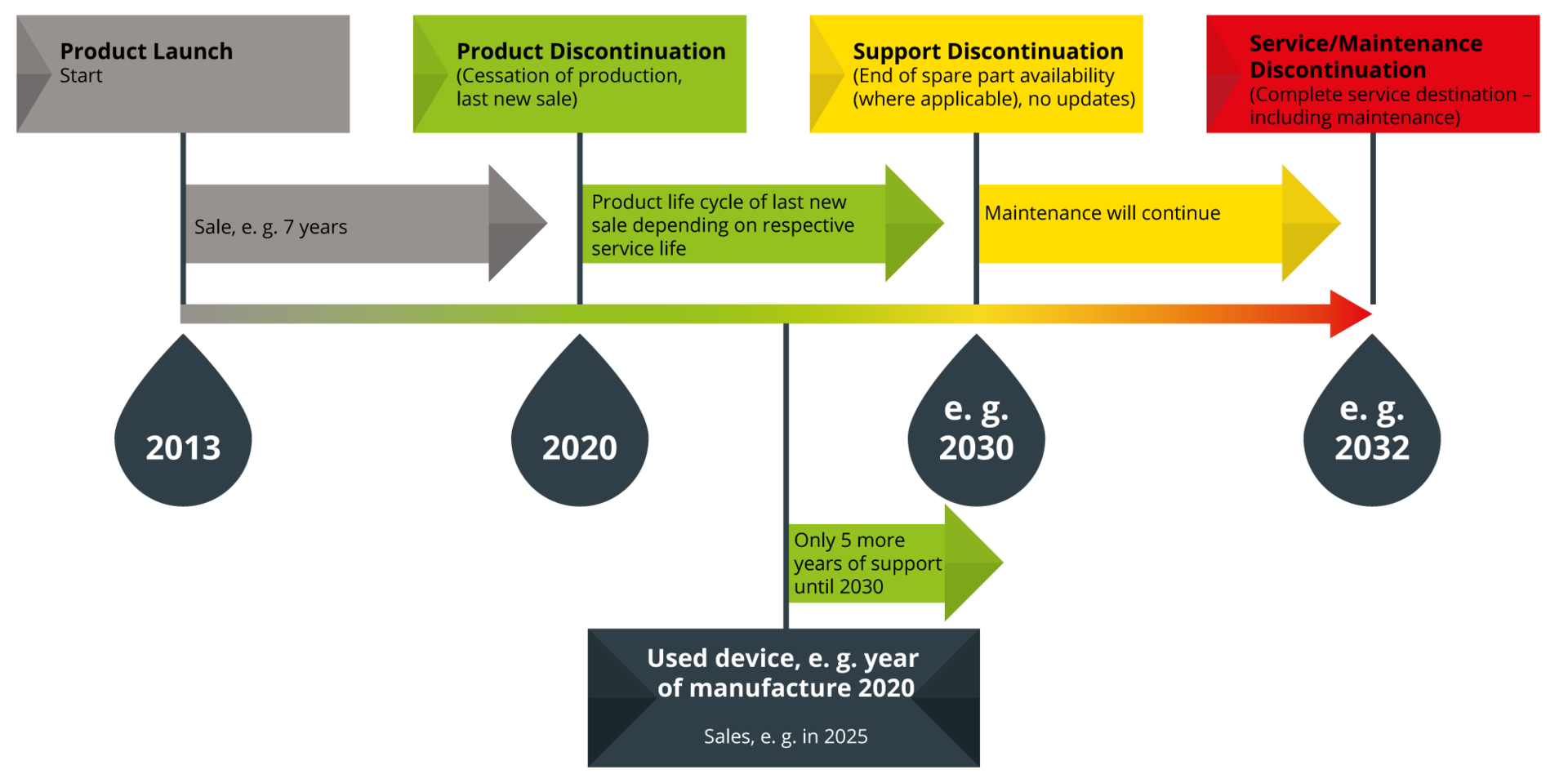
Example: You purchase a used device in 2025 that was manufactured in 2020. According to our lifecycle traffic light system, support for this device continues until 2030, up to the support discontinuation stage.
- The product is no longer manufactured or sold as new.
- It continues to be supported; spare parts are available until at least the end of its service life.
- Maintenance can be carried out as usual.
- The product may still receive updates or major updates.
- For distributed products: the manufacturer discontinues the product, and proxomed communicates this on their behalf, or the product is no longer distributed by proxomed and customers are referred directly to the manufacturer.
- The service life of the last sold product has expired.
- Spare parts are limited or no longer available.
- Maintenance can still be performed; however, functionality may be affected due to wear or hidden defects.
- The product no longer receives updates or major updates (exceptions: safety-relevant updates).