

Del lado seguro – Con un sistema completo de aseguramiento de la calidad
Nuestra empresa cuenta con un sistema de gestión de calidad para dispositivos médicos conforme a la norma EN ISO 13485, y cumple además con los requisitos del Reglamento Europeo de Dispositivos Médicos (MDR), declarando la conformidad de sus productos de acuerdo con dicho reglamento.
Nuestro compromiso: por una sociedad saludable y por su seguridad legal.




Son todos los productos utilizados con un propósito médico, como diagnóstico, tratamiento o alivio de enfermedades.
Entre ellos se incluyen equipos de ultrasonido, endoscopios, implantes y equipos de entrenamiento utilizados en terapia y rehabilitación.
Estos productos se caracterizan por su precisión comprobada, seguridad técnica y biológica, eficacia y facilidad de uso.
A ellos se les aplican los más altos estándares y requisitos.
Los requisitos son extremadamente exigentes.
Según el Reglamento Europeo de Dispositivos Médicos (UE 2017/745) y un sistema de gestión de calidad certificado conforme a EN ISO 13485, todas las etapas de planificación, desarrollo y producción son planificadas, implementadas, verificadas y validadas de manera sistemática por un equipo de expertos altamente calificados y motivados, utilizando normas internacionales.
Estos requisitos también se aplican a fabricantes con sede fuera de la Unión Europea, si comercializan dispositivos médicos dentro de la UE.
Cada año, organismos independientes (en nuestro caso TÜV Süd) realizan auditorías de supervisión para evaluar el cumplimiento de los estándares de calidad establecidos./p>
Esto puede verificarse en la Declaración de Conformidad del producto.
En ella, el fabricante indica qué requisitos legales cumple el producto —como mínimo, el Reglamento Europeo de Dispositivos Médicos (2017/745 – MDR).
El fabricante o proveedor del equipo está obligado a entregar esta declaración cuando se le solicite. Cada institución sanitaria debe conservar dichas declaraciones para evitar posibles consecuencias legales ante una inspección de las autoridades competentes.
Cada dispositivo debe mostrar su conformidad mediante el marcado CE, que normalmente se encuentra en la placa de identificación.
En muchos dispositivos médicos de clase superior, el número de identificación de cuatro dígitos del organismo notificado (por ejemplo, TÜV Süd – CE 0123) debe figurar junto al marcado CE.
Existen dispositivos médicos destinados a diferentes grupos de usuarios, definidos por el fabricante según su uso previsto.
Algunos dispositivos están diseñados especialmente para uso doméstico o por parte de personas no profesionales.
En el caso de los dispositivos destinados a uso profesional, la ley establece que solo terapeutas o profesionales capacitados y debidamente instruidos en el uso del equipo médico pueden realizar diagnósticos o terapias.
Los equipos de entrenamiento certificados como dispositivos médicos cuentan con:
- Eficacia clínica comprobada científicamente,
- Gestión de riesgos integral,
- Seguridad biológica, eléctrica y mecánica verificada.
Además, estos productos están sujetos a una supervisión técnica y clínica continua durante su uso, con el fin de minimizar los riesgos y garantizar el éxito terapéutico —durante toda su vida útil.
Los dispositivos médicos están sujetos a inspecciones de seguridad y/o metrológicas periódicas, definidas por el fabricante.
Las evidencias de estas inspecciones deben presentarse a las autoridades de salud cuando lo soliciten.
Al firmar un contrato de mantenimiento, estas revisiones pueden realizarse de forma automatizada y quedar debidamente documentadas.
Importante: conectar uno de nuestros dispositivos médicos a accesorios o sistemas de control no autorizados por nosotros puede provocar la pérdida de la conformidad del producto, transfiriendo toda la responsabilidad legal al operador.
Cualquier modificación en un equipo instalado —por ejemplo, la instalación de software de terceros o el cambio de pantalla— debe ser autorizada por escrito por el fabricante. De lo contrario, el equipo pierde su certificación de seguridad.
En caso de daños o lesiones, el operador asume toda la responsabilidad legal, y se anulan las garantías y derechos de reclamación.
Desde hace más de 30 años, nuestros clientes en todo el mundo confían en nuestros sistemas médicos de prueba, entrenamiento y terapia, apoyando nuestra visión de una sociedad saludable.
Con nosotros, usted está del lado seguro: PHYSIOMED, como empresa de tecnología médica certificada bajo EN ISO 13485, garantiza los más altos estándares de seguridad y calidad. Nuestros productos cumplen con el Reglamento Europeo de Dispositivos Médicos (MDR) y llevan el marcado CE.
Como empresa certificada, definimos una vida útil para nuestros productos, durante la cual —después de su comercialización— el dispositivo no representa ningún riesgo para pacientes ni usuarios.
Durante este periodo, el fabricante está obligado a ofrecer repuestos originales para posibles reparaciones, de acuerdo con el diseño y la gestión de riesgos del producto.
Además, realizamos la vigilancia del mercado obligatoria durante toda la vida útil del producto.
Dentro del ciclo de vida del dispositivo, también se debe considerar la fase de discontinuación. Nuestro objetivo es ofrecer a nuestros clientes un soporte prolongado y profesional, a través de nuestro sistema de semáforo, que aplicamos tanto a nuestros productos propios como a los que distribuimos.
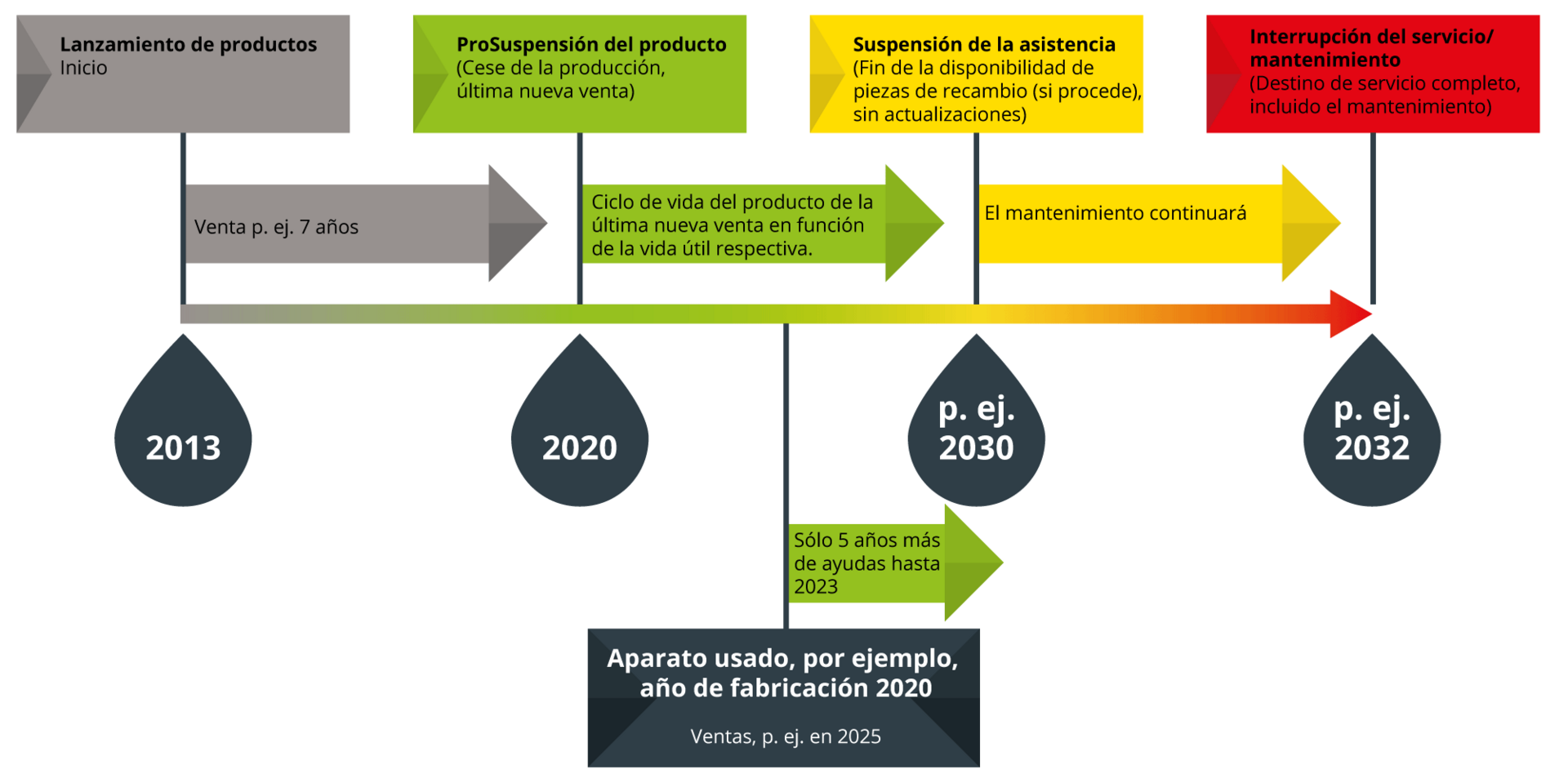
Ejemplo: usted adquiere en 2025 un equipo usado, fabricado en 2020. Según nuestro sistema de ciclo de vida (semáforo), el soporte para este equipo continuará hasta 2030, fecha en la que finaliza el soporte técnico.
- El producto deja de fabricarse, no se realizan nuevas ventas.
- El producto sigue siendo objeto del servicio de asistencia y se dispondrá de piezas de repuesto al menos hasta el final de su vida útil.
- El mantenimiento continúa con normalidad.
- El producto puede seguir recibiendo todo tipo de actualizaciones.
- Productos de otros fabricantes: El fabricante suspende el producto, que es asumido por proxomed en su nombre, o el producto deja de venderse a través de proxomed y el cliente es remitido al fabricante.
- La vida útil del último producto vendido ha llegado a su fin.
- No hay piezas de repuesto disponibles, o solo de forma limitada.
- El mantenimiento puede continuar. Nota: Se intentará preservar la funcionalidad del sistema, a pesar de su antigüedad y del posible desgaste causado por defectos mecánicos y eléctricos ocultos.
- El producto ya no recibe actualizaciones/actualizaciones importantes (a excepción de las actualizaciones de seguridad).
- Por ejemplo, el producto presenta defectos técnicos (por ejemplo, las cubiertas se rompen al retirarlas), o el producto ya no se corresponde con el estado actual de la técnica.
- Ya no se dispone de técnicos cualificados formados para llevar a cabo el mantenimiento de acuerdo con la MPG.
- Ya no se realiza el mantenimiento.
- El producto ya no recibe actualizaciones/actualizaciones importantes.